In the fast-moving world of medical diagnostics, it’s really important for both manufacturers and healthcare providers to understand the rules and standards around products labeled 'For In Vitro Diagnostic Use Only' (or IVD). I mean, as MarketsandMarkets reports, the global IVD market is expected to hit around $85 billion by 2025 — that’s mostly because people are craving more accurate and quicker diagnostic tests.
One standout company in this space is Zhejiang Pushkang Biotechnology Co., Ltd. — they started back in 2014 and have been leading the way with innovative tools. They focus heavily on R&D, making and promoting IVD products. What’s cool about Pushkang is their use of centrifugal microfluidics technology, which they've used to create a bunch of point-of-care testing (POCT) solutions for things like blood coagulation, biochemistry, CLIA, and molecular diagnostics.
And no matter what, they stick closely to strict regulations and industry standards to be sure their products are safe and reliable. Basically, Pushkang’s all about pushing healthcare forward worldwide.
Understanding the Basics of In Vitro Diagnostic Regulations
In vitro diagnostics, or IVDs for short, are such a crucial part of today’s healthcare scene. They’re practically lifesavers when it comes to catching, preventing, and managing diseases. If you’re in the game—whether you're a manufacturer or a healthcare pro—it's pretty important to get a handle on the basic regulations surrounding IVDs. The FDA, for instance, sorts these devices into different classes based on how risky they are. The lower-risk ones—Class I—just need to follow some general controls. But the more advanced and riskier devices, like Class II and III, have to go through tougher premarket approvals before hitting the market. According to a report from Grand View Research, the global IVD market is expected to hit about $87.7 billion by 2027—so, it's a massive and rapidly growing field. That just underscores why sticking to the rules isn’t just good practice, but absolutely essential if you want to stay ahead.
Beyond that, following standards set by organizations like ISO—particularly ISO 13485—is super important. This particular standard lays out what’s needed for quality management in designing and making medical devices, including IVDs. It’s worth noting that a study from Research and Markets found that around 60% of IVD manufacturers saw better efficiency and easier market access after adopting ISO standards. As the industry keeps growing and everyone demands more reliable diagnostic tools, understanding and following these regulations isn’t just smart—it’s vital if you want your success to stick around.
In Vitro Diagnostic Regulations Compliance Overview
This chart provides a visual representation of the number of in vitro diagnostic products classified under different regulatory classes. The data highlights the distribution of products, showing that Class II devices are the most prevalent among the categories.
Key Differences Between FDA and EU Regulations for Diagnostics
Getting a good grasp of the regulatory scene for in vitro diagnostics is pretty important, especially when you're trying to compare what the FDA and EU have going on. From recent analyses, it’s clear that while both agencies are out there to make sure products are safe and work well, they don’t always do things the same way. For example, the FDA’s 510(k) process lets companies get their devices to market more quickly if they can show it’s pretty much the same as something already approved. On the flip side, the EU’s CE marking process tends to require a lot more detailed clinical data from the start, which can make things take a bit longer.

As diagnostics technology keeps evolving, we’re seeing a real push for better policies—especially when it comes to companion diagnostics, or CDx. A side-by-side look at how agencies like the FDA and the EMA handle regulations really points to the need for better teamwork and maybe even some global standards, especially since new tech like AI is starting to make its way into clinical tools. Those AI applications, especially in digital pathology, promise to change everything about how we diagnose, but they also bring new hurdles for regulators to figure out.
Some tips:
- Make sure you stay up-to-date with the latest rules and guidance from both the FDA and EU so you’re not caught off guard.
- And it’s usually a good idea to chat with regulatory bodies early on during product development. Getting their feedback sooner rather than later can really smooth out the approval process and help you get to market faster.
Comparative Analysis of IVD Standards: ISO vs. CLIA
When you're working in the world of in vitro diagnostics (IVD), making sure you're compliant with all the regulations and standards is super important for keeping your products safe and effective. There are two main sets of guidelines you need to be familiar with: the International Organization for Standardization (ISO) and the Clinical Laboratory Improvement Amendments (CLIA). Both aim to keep diagnostic tests quality high, but they approach things quite differently. ISO standards are recognized worldwide and mostly focus on creating best practices for manufacturing and quality management—that kind of stuff. On the other hand, CLIA is specific to the U.S. and puts a big emphasis on making sure lab tests are accurate and reliable.
Tip: If you’re developing an IVD product, it’s a smart move to get a good grip on both ISO and CLIA regulations early on. Knowing the little differences between them can really help speed up your development process and get you to market faster.
Plus, ISO standards tend to be a bit more flexible—they allow for some interpretation depending on your particular IVD. That can be a real bonus if you're looking to innovate. Meanwhile, CLIA is a bit more strict, with clear-cut rules for how labs should operate, mainly to ensure quality and consistency.
Tip: Keep an eye on updates and changes in both ISO and CLIA by attending webinars or reading industry magazines. Staying in the loop is key—being proactive with regulations means your team will be prepared to handle any adjustments smoothly.
Navigating Compliance Challenges in In Vitro Diagnostics
Dealing with compliance issues in in vitro diagnostics (IVDs) can be pretty challenging, especially for companies like Zhejiang Pushkang Biotechnology Co., Ltd., which is really pushing the envelope when it comes to innovative diagnostic solutions. Since starting out in 2014, PUSHKANG has made a name for itself as a high-tech company that focuses on a variety of IVD products. They leverage advanced centrifugal microfluidics technology, making it possible to perform quick and reliable tests for things like blood coagulation, biochemistry, CLIA, and molecular diagnostics. All of this aligns with industry standards that stress how vital accuracy and regulatory compliance are.
If you want to stay ahead in this game, it’s so important to really understand the rules set by authorities like the FDA and EMA. According to a report from Market Research Future, the global IVD market is projected to hit about $85 billion by 2025—that’s a clear sign that there’s rising demand for products that are both top-quality and compliant. Following these strict regulations isn’t just about avoiding penalties; it’s also about ensuring patient safety and building trust in your products’ effectiveness. For companies like PUSHKANG, that means keeping a close eye on regulatory updates and being ready to adapt quickly—otherwise, they risk running into issues with quality assurance, clinical validation, and breaking into new markets in a pretty competitive scene.
The Role of Quality Management Systems in IVD Manufacturing
You know, quality management systems (or QMS for short) are absolutely essential when it comes to making in vitro diagnostics (IVD). As rules and standards keep shifting, companies really gotta stay on top of their game and make sure their processes are current and compliant. Actually, the global market for IVD quality control—think of it like the quality side of things—hit around USD 1.3 billion in 2023, and it’s expected to grow by about 4.2% annually over the next few years. That’s a pretty clear sign that having solid QMS setups—especially ones that stick to international standards like IVDR and Indian MDR—is more important than ever.
Lately, we’re seeing a lot of companies jumping on modern QMS solutions to make their quality assurance and manufacturing smoother. For example, many are now using cutting-edge tech platforms that help them juggle compliance effortlessly across different markets, like China and the US. With all these changes, stakeholders are facing the tricky task of understanding and adapting to new FDA quality regulations. The goal is to make sure their IVD products are safe, perform well, and are efficient. Overall, sticking to high standards isn’t just about ticking boxes for regulators; it’s also about staying competitive in an ever-changing market—and honestly, that’s what really matters in the end.
Comprehensive Guide to Understanding For In Vitro Diagnostic Use Only Regulations and Standards - The Role of Quality Management Systems in IVD Manufacturing
| Regulation/Standard |
Description |
Importance |
Quality Management System Role |
| ISO 13485 |
International standard specifying requirements for a quality management system where an organization needs to demonstrate its ability to provide medical devices and related services. |
Ensures consistent quality and safety of medical devices. |
Provides a structured framework for managing quality throughout the IVD product lifecycle. |
| FDA 21 CFR Part 820 |
U.S. regulation for quality system requirements for manufacturers of medical devices. |
Regulates the quality management systems to ensure safety and efficacy of diagnostic products. |
Outlines necessary quality control processes in the design and production of IVD products. |
| IVDR (EU) 2017/746 |
European In Vitro Diagnostic Regulation that governs the marketing and use of IVD medical devices. |
Enhances patient safety and ensures performance of IVD devices within the EU market. |
Enforces rigorous pre-market and post-market compliance measures for quality assurance. |
| CLIA |
Clinical Laboratory Improvement Amendments that regulate laboratory testing in the U.S. |
Ensures the accuracy and reliability of laboratory tests. |
Promotes continuous quality improvement in laboratory practices for IVDs. |
Future Trends in In Vitro Diagnostic Regulations and Standards
Looking ahead, the world of in vitro diagnostics (or IVD for short) is really set for some big changes, thanks to new trends catching on and updates to regulations that are shaking things up. The global IVD market is expected to hit around USD 149.4 billion by 2032 – that’s a pretty huge number! What’s driving this growth? A big part of it is the integration of cutting-edge technologies. Digital diagnostics, which blend data analysis with the usual lab tests, are totally changing the game. They're making disease detection more precise and paving the way for personalized medicine — you know, treatments tailored just for each patient. It’s not just about smarter insights; it also makes the whole diagnostic process smoother, giving healthcare providers better tools to care for their patients in a more personal way.
On the regulatory side of things, there’s a major shake-up coming in Great Britain. Starting June 16, 2025, they’ll roll out a whole new set of rules for medical devices. Things will get stricter, especially when it comes to post-market surveillance — basically, manufacturers will need to keep a closer eye on their products’ safety and performance after they’ve been released. These changes are super important because they help make sure that new IVD solutions can be safely introduced into everyday clinical practice. As everyone in the industry gets used to these new standards, it’ll be really important for all the different players — companies, regulators, and healthcare providers — to work together. Only then can we keep pushing innovation forward while still making sure everything stays compliant and safe in this fast-changing field.

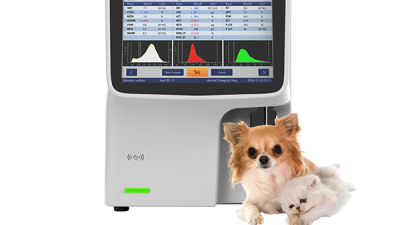
Enhancing Animal Care with Precision: Exploring the Benefits of the Veterinary Hematology Analyzer MX30V
Enhancing animal care has always been a top priority in veterinary medicine, and the Veterinary Hematology Analyzer MX30V is setting new standards in this area. This innovative device is specifically designed for veterinary diagnostic laboratories, enabling precise identification of subtle changes in complete blood counts (CBC). The analyzer boasts a user-friendly interface with its 10.4 inch TFT touch screen, making it accessible for veterinary professionals at all levels of expertise.
One of the standout features of the MX30V is its economy and efficiency; it requires only a minimal sample volume of 9μl of whole blood per test. This not only ensures that more tests can be conducted with less blood drawn from the animal but also facilitates a quick turnaround time, with results available in just one minute. Furthermore, the built-in thermal printer and the customizable, user-defined animal species settings enhance the flexibility and functionality of the device, making it a valuable tool in any veterinary practice.
Reliability and accuracy are critical in diagnosing health issues, and the MX30V shines in this regard as well. With its automatic calibration and comprehensive quality control programs, veterinary professionals can trust the results they receive. Additionally, the operational simplicity of “one-click testing” and the original cyanide-free reagents support a safe, biohazard-free working environment, further emphasizing the MX30V's role in enhancing animal care with precision.
FAQS
: IVDs are critical tools for disease detection, prevention, and management in modern healthcare.
The FDA classifies IVDs based on risk: Class I devices pose the least risk with general controls, while Class II and Class III devices require more stringent premarket submissions and approvals.
Compliance with ISO standards, particularly ISO 13485, helps improve efficiencies and market access for IVD manufacturers.
The FDA's 510(k) process allows faster market entry for substantially equivalent devices, whereas the EU's CE marking requires more comprehensive clinical data, leading to longer approval times.
New AI applications in digital pathology could revolutionize diagnostics but also present challenges in regulatory oversight, highlighting the need for refined policies.
Staying abreast of the latest updates from FDA and EU guidance documents ensures compliance with regional regulations and facilitates smoother product approval and market entry.
IVD companies must address challenges related to quality assurance, clinical validation, and market access, all while adapting to changing regulations.
The global IVD market is projected to grow significantly, reaching around USD 87.7 billion by 2027, emphasizing the need for compliance with regulatory standards.
Early engagement with regulatory agencies can facilitate smoother product approvals and market entry by addressing compliance issues proactively.
Conclusion
In the fast-changing world of in vitro diagnostics (IVD), it's so important for both manufacturers and healthcare providers to really get a handle on the rules and standards, especially when it comes to products labeled 'For In Vitro Diagnostic Use Only.' This guide is pretty much your starting point—it breaks down the basics of IVD regulations and points out the main differences between the FDA and European Union rules. Plus, it takes a closer look at standards like ISO and CLIA, helping companies understand what hurdles they might face when making sure they’re compliant.
Quality Management Systems are also a big deal here—they basically make sure the products are trustworthy and safe. As the industry keeps evolving, it’s crucial for companies like Zhejiang Pushkang Biotechnology Co., Ltd., which is creating innovative diagnostic tools, to stay on top of future regulatory trends. By understanding these regulations and standards, PUSHKANG can confidently move forward, contributing to cutting-edge diagnostic solutions while sticking to the tough requirements for 'For In Vitro Diagnostic Use Only' products.